Overview¶
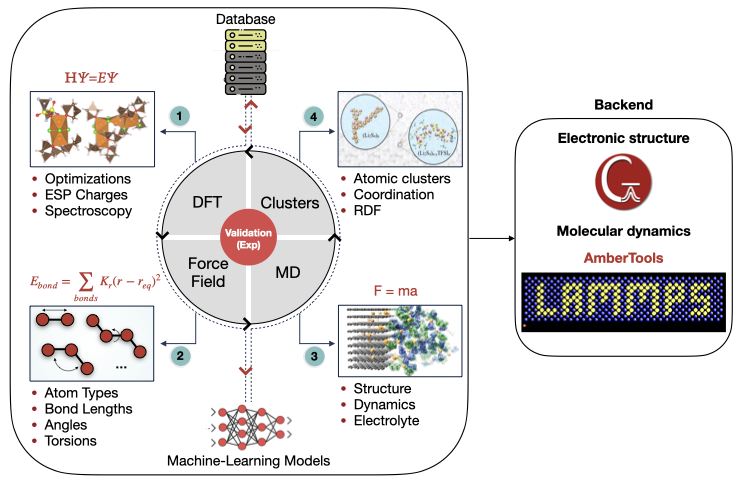
MISPR (Materials Informatics for Structure-Property Relationships) is a high-throughput computational infrastructure aimed at guiding and accelerating materials discovery, optimization, and deployment for liquid solutions by seamlessly integrating density functional theory (DFT) with classical molecular dynamics (MD) techniques.
MISPR is motivated by the Materials Genome Initiative (MGI) principles and is built on top of open-source Python packages developed for the Materials Project such as pymatgen, FireWorks , and custodian, as well as MDPropTools, which is an in-house package for analyzing MD output and trajectory files.
Features of MISPR include:
Automation of DFT and MD simulations and all their underlying tasks from file management and job submission to supercomputing resources, to output parsing and data analytics; a task that can be done to a single molecule/system or to a large number of systems in parallel
Creation of computational databases of force field parameters and DFT and MD derived properties of molecular systems for establishing structure-property relations and maintaining data provenance and reproducibility
Detection of the inevitable errors that occur during the simulations and their on-the-fly correction based on template responses that have been designed relying on human intuition coupled with extensive experience to significantly improve the success rate of high-throughput simulations while eliminating human intervention
Support for flexible and well-tested DFT workflows that compute various properties of individual molecular species or complexes such as bond dissociation energy, binding energy, redox potential, and nuclear magnetic resonance (NMR) tensors
Derivation of many molecular ensemble properties such as radial distribution functions, diffusion coefficients, viscosity, and conductivity of liquid solutions, which are critical to understanding complex inter- and intra-atomic interactions controlling the performance of solutions within various chemistry, biology, and materials science applications
Seamless integration of DFT and MD simulations through hybrid workflows that enable force field generation and information flow between the two length scales to allow exploring wide chemical and parameter spaces (e.g., temperature, pressure, concentration, etc.), a task that can be infeasible experimentally and challenging using manual calculations
Automatic extraction of hundreds of thousands of solvation structures from MD ensembles and their use in DFT workflows to accurately represent the electronic environment, which is crucial to derive reliable energetics and other properties such as NMR chemical shifts and redox potentials and match them to experimental data